
The Silent Saboteur: how leaky Cre shapes (and skews) in vivo immunology
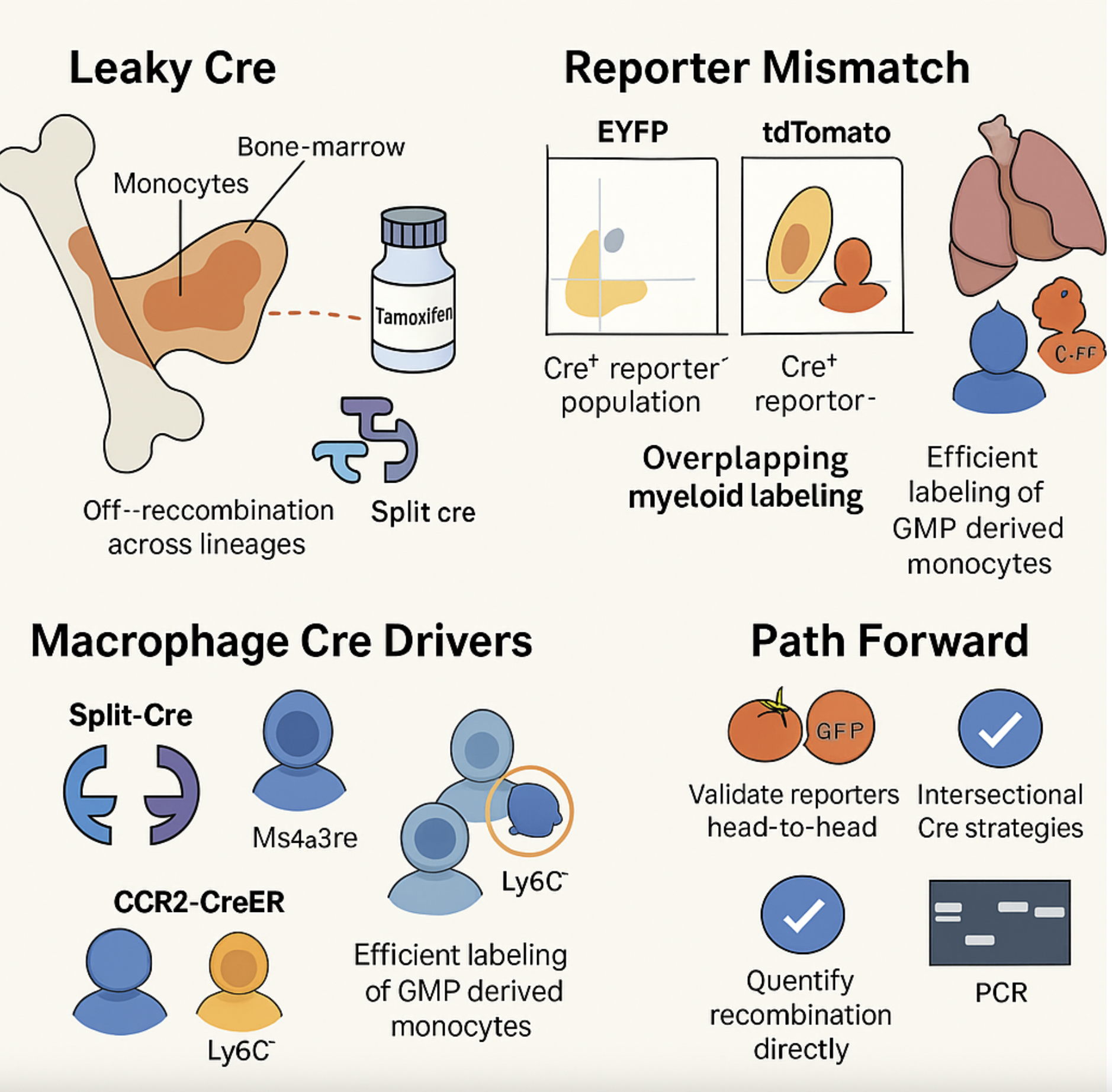
One of the major technical bottlenecks in doing rigorous in vivo immunology with mouse Cre/lox systems is “leaky Cre” activity defined as the recombination that occurs in the wrong cells, at the wrong time, or even in the absence of intentional induction, silently scrambling the very compartments we think we are manipulating. Since Orkin’s 1999 paper in PNAS, we’ve known of Cre activity in unexpected lineages or developmental windows (PNAS). Over the years increasingly sensitive indicators have revealed ectopic, germline, and tamoxifen-independent recombination across many ostensibly tissue- or time-restricted Cre and CreERT2 lines (Blank, Palmer and Tallquist). Against that backdrop, this new work on leaky or under-reporting Cre in T cells (by Kansas) should be read as part of a larger cautionary story: in myeloid and macrophage biology as much as in lymphocytes, the fidelity of our conclusions is often limited less by the elegance of our hypotheses than by the quiet, cumulative consequences of imperfect Cre control.
In Kansas’s paper - a widely used Rosa26 EYFP Cre-reporter line (R26^EYFP, JAX 007903) substantially under-reports Cre activity in T cells compared with an otherwise identical tdTomato reporter (R26^tdTomato, JAX 007914) when driven by distal Lck–Cre (dLcK-Cre). In dlck-Cre × R26^EYFP/tdTomato mice, CD4⁺ and CD8⁺ single-positive thymocytes and peripheral naïve T cells segregated into two major Cre⁺ populations: EYFP⁺tdTomato⁺ and tdTomato⁺EYFP⁻, with virtually no EYFP⁺tdTomato⁻ cells. The tdTomato-only population was consistently present in all mice and in both CD4 and CD8 lineages, indicating that a substantial fraction of bona fide Cre-expressing T cells never turn on EYFP, despite identical locus and regulatory elements. EYFP and tdTomato patterns in earlier thymic stages and in B cells and NK cells matched prior dlck-Cre characterization, confirming that the discordance reflects loss of EYFP detection rather than ectopic tdTomato expression. The author concludes that this EYFP reporter (and likely similar EYFP-based Rosa26 constructs) fails to label a sizeable subset of Cre⁺ cells, with major implications for lineage tracing, use of EYFP⁻ cells as “internal Cre⁻ controls,” and interpretation of inducible or asynchronous Cre systems.
These results echo the persistent challenges inherent to macrophage- and monocyte-specific Cre lines: none offers both full penetrance and absolute specificity. Instead, drivers such as LysM-Cre, Cx3cr1-Cre/CreER, Csf1r-Mer-iCre-Mer, and CD11b-Cre display overlapping myeloid labeling, tissue- and ontogeny-dependent variability, and recombination efficiencies that fluctuate with locus and induction (Ren, Williams groups and write-up by Jakubzick & Janssen group). We talk about this a little bit in my recent review in elife as well (Damani-Yokota & Khanna). In this context, the binary “split-Cre” strategy pioneered by Steffen Jung’s group, in which N- and C-terminal Cre fragments are driven by distinct promoters (e.g., Sall1^nCre × Cx3cr1^cCre or Lyve1^nCre × Cx3cr1^cCre), has emerged as an elegant way to intersectionally target microglia and CNS border-associated macrophages with exceptional specificity, albeit at the predictable cost of somewhat reduced overall recombination efficiency compared with conventional single-promoter Cre lines (Jung).
Florent Ginhoux’s monocyte/macrophage fate-mapping Ms4a3Cre and Ms4a3CreERT2 lines, originally described (in Cell) , label granulocyte–monocyte progenitors and their monocyte/granulocyte progeny with high efficiency while sparing lymphocytes and most tissue DCs, enabling precise quantification of monocyte contributions to resident macrophage pools across tissues and inflammation models. These Ms4a3-based models have since been deployed to dissect monocyte fates in pancreatic cancer, cochlear macrophage ontogeny , and brain-infiltrating macrophages after injury, often in combination with scRNA-seq and multi-omics (Ginhoux, Okano, Ginhoux & Okano, and Rossi groups). Parallelly, CCR2-CreER mice as a tamoxifen-inducible monocyte-tracing line, widely used to track Ly6C^hi/lo monocytes and their progeny in brain microglia, border-associated macrophages, and other tissues (Kuan group). Despite being powerful systems, they come with known blind spots: Ms4a3 labels GMP-derived monocytes but not embryonic progenitors, CCR2 misses CCR2⁻ monocyte compartments and can label some non-macrophage myeloid cells, and both depend on reporter alleles whose recombination efficiency and expression thresholds vary (Williams, Smith and Ren groups).
In our research as macrophage biologists that are ardently establishing systems for specific depletion of nerve associated CD169+ macrophages (Khanna group and NAMs), the cre-lox system and the inherent flaws are critical. A pragmatic path forward, in light of this EYFP–tdTomato discordance and the imperfections of macrophage Cre systems, is to treat any single Cre–reporter combination as a hypothesis generator rather than ground truth. I am still learning the system and there are others that are far more advanced in their use and understanding of these systems. With all this in mind, some things that I (and many others) have been thinking about are conceptual and technical steps that need to be taken henceforth:
(i) favoring brighter, high-dynamic-range reporters like tdTomato (or dual fluorophores) and validating them head-to-head when possible;
(ii) combining complementary drivers (e.g., Ms4a3Cre/CreERT2 plus CCR2-CreER, or Csf1r-Mer-iCre-Mer) to bracket the monocyte- versus embryonic-derived macrophage space;
(iii) using intersectional strategies (e.g., Cre/Dre or Cre/Flp systems such as CD45-Dre × Gata6-CreER for cavity macrophages) to restrict labeling to defined ontogeny–tissue niches; and
(iv) quantifying recombination directly at both the reporter and the floxed allele by flow, PCR, or single-cell multi-omics in each tissue and time point of interest.
Maybe If we standardize these validation steps and report them transparently, we can still obtain robust, reproducible, and biologically meaningful insights from monocyte/macrophage Cre systems even while fully acknowledging that every current tool is, by design, an imperfect lens on myeloid biology.
Generated using ChatGPT - a graphical abstract of points discussed in this article.

